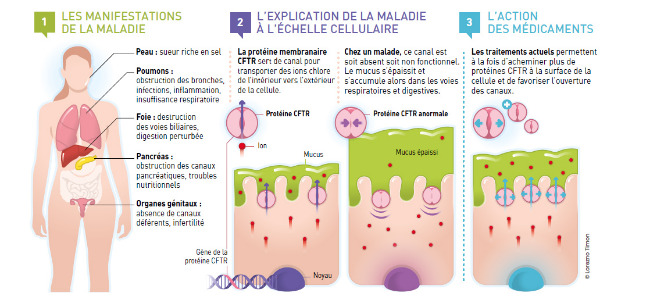
Histoire de la description de la maladie

Dorothy Hansine Anderson fut la première à décrire la mucoviscidose en tant que maladie à part entière.
La mucoviscidose, décrite scientifiquement comme maladie en 1936, était en fait déjà connue depuis longtemps.
Selon Busch, une des premières descriptions médicales des lésions pancréatiques rencontrées dans la mucoviscidose pourrait être un rapport d'autopsie fait par le professeur Pieter Pauw, à Leyde aux Pays-Bas en 1595,, dans lequel il décrit une patiente chétive de 11 ans supposée ensorcelée et présentant un pancréas élargi, dur et blanc.
Au XIXe siècle, le médecin viennois Karel Rokitansky signale chez un foetus un cas fatal de péritonire méconiale, qui sera plus tard identifié comme une complication de l'iléus méconial, obstruction intestinale néonatale présente chez certains enfants mucoviscidosiques décrite en 1905 par Landsteiner.
Au début du XXe siècle apparaissent les premières observations associant maladie pulmonaire, diarrhée et anomalie pancréatique avec plusieurs cas dans la même famille.
En 1912, Garrod décrit des familles dont les enfants présentent une diarrhée graisseuse et meurent d'infection pulmonaire. Ces descriptions se focalisent le plus souvent sur les problèmes digestifs, stéatorrhée et troubles pancréatiques, et leurs auteurs en font une forme de maladie coeliaque même si les problèmes bronchopulmonaires sont également notés.
C'est en 1936, dans une thèse écrite en allemand et présidée par le pédiatre suisse Guido Fanconi, que la maladie est décrite pour la première fois chez des enfants supposés atteints de maladie coelique, sous le nom de « fibrose kystique du pancréas et bronchectasie ».
La mucoviscidose ne fut considérée comme une entité pathologique distincte qu'en 1938 par la pédiatre américaine Dorothy Hansine Anderson, médecin au Babies' Hospital de New York, qui publia un article intitulé « Fibrose kystique du pancréas et ses relations avec la maladie cœliaque ». C'est en pratiquant des autopsies sur des nourrissons qu'elle décrivit les caractéristiques cliniques et histologiques de la maladie, notamment l'obstruction intestinale néonatale, les complications respiratoires et digestives et les lésions histologiques spécifiques du pancréas. Elle relia cette maladie à un déficit en vitamine A et persista à soutenir cette théorie pendant de nombreuses années bien qu'elle ne fût jamais confirmée.
Le terme de mucoviscidosis, créé à partir des termes « mucus » et « visqueux », fut utilisé pour la première fois en 1943 par le docteur Sydney Farber médecin-chef au Children's Hospital de Boston, afin de corriger la dénomination employée par Dorothy Andersen, centrée sur le pancréas. Farber était persuadé que la maladie était due à une diffusion généralisée de mucus visqueux. Le terme de mucoviscidosis reste très employé dans le monde, et notamment en France, et est parfois préféré au terme anglais cystic fibrosis26.
Le caractère héréditaire et le mode de transmission récessif furent suggérés en 1945 par Dorothy Andersen et Hodges.
C'est à la suite d'une vague de chaleur entraînant un état de prostration chez de jeunes patients del'hôpital Columbia de New York en 1948 que le docteur Paul di Sant' Agnese découvre et décrit en 1953 les anomalies électrolytiques dans la sueur des malades (augmentation importante du chlore, du sodium et moins marquée du potassium) , permettant d'envisager un diagnostic spécifique à la maladie : le test de la sueur..
Le recueil de la sueur était effectué grâce à des buvards. De réalisation difficile, la technique fut par la suite simplifiée, Le test de la sueur devint et reste jusqu'à ce jour le test le plus fiable pour établir le diagnostic, en dehors de l'analyse génétique qui ne fut disponible que bien plus tard.
C'est au début des années 1980 que le lien physiopathologique fut fait entre d'une part l'anomalie de la sécrétion de mucus, entraînant des obstructions glandulaires avec anomalies histologiques et d'autre part l'anomalie de la sueur, entraînant des sécrétions salées sans anomalie histologique.
En 1981, Knowles découvrirent que le potentiel électrique au niveau de la muqueuse nasale des patients mucoviscidosiques étaient plus électronégatifs que chez les sujets sains, ce qu'ils expliquèrent par une réabsorption massive du sodium entraînant une déshydratation à la surface de l'épithélium. Cette découverte faisait le lien physiologique entre les poumons,, le panccréas et les glandes sudoripares. Le lien commun expliquant l'atteinte des différents organes n'était pas le mucus lui-même, mais les anomalies électrolytiques.
En 1983 Quinton, lui-même atteint de la mucoviscidose, montra que l'imperméabilité au chlore qu'il avait trouvée dans les glandes sudoripares était la cause de l'augmentation des électrolytes dans la sueur. Ceci fut considéré comme une étape majeure dans la compréhension de la maladie.
Restait à localiser et à identifier le gène dont la mutation provoque la mucoviscidose, une tâche rendue difficile car seules la sémiologie clinique, le mode de transmission autosomique récessif et l'anomalie de transport du chlore étaient alors connus.
En l'absence de toute connaissance sur la protéine défectueuse, et donc de marqueur chimique identifié, la récente technique du clonage positionnel - ou génétique inverse, qui permet d'identifier un gène sans connaître la protéine qu'il code, fut utilisée. Cette méthode consiste grâce à des analyses statistiques de liaison génétiques à déterminer la région du chromosome où le gène a une forte probabilité de se trouver, afin de la séquencer et d'étudier les gènes exprimés. Le gène de la mucoviscidose est le premier gène à avoir été cloné uniquement par analyse de liaisons.
En 1985, Eiberg trouvent un lien entre l'enzyme paraoxinase (PON) et le gène CF en étudiant des familles où il y avait plus d'un enfant atteint. La même année, Tsui, à la suite d'études sur des souris hybrides, localisent le gène sur le bras long du chromosome 7 grâce à un marqueur RLFP lié à la fois à la paraoxinase et à la mucoviscidose.
En 1989, le gène impliqué dans la mucoviscidose est isolé par les équipes de Lap-Chi Tsui, Collins et Riordan. L'anomalie génétique à l'origine de la maladie est enfin découverte, il s'agit d'une mutation d'un gène localisé en 7q31 et contenant 27 exons, nommé cystic fibrosis (CF) codant une protéine transmembranaire appelée cystic fibrosis transmenbrane conductance regulator (CFTR) composée de 1480 acides aminés.
Ce n'est qu'un peu plus tard qu'on apporta les preuves que CFTR était bien un canal du chlore,La découverte de l'anomalie génétique permit par la suite d'ajouter le génotypage au protocole diagnostique, et d'envisager la thérapie génique.