C'est un grand gène constitué d'environ 250 000 paires de bases. Il est localisé sur le locus 7q31.2, dans la région q31.2 du bras long du chromosome 7.
Les Mutations du gène CFTR
On dénombre en 2011 plus de 1900 mutations du gène CFTR.
Les mutations du gène CFTR sont regroupées en 6 classes en fonction des conséquences fonctionnelles qu'elles occasionnent.
Certaines mutations entraînent des anomalies quantitatives ou qualitatives sur la protéine CFTR :
-
Classe 1 : mutations altérant la production de la protéine CFTR.
-
Classe 2 : mutations perturbant le processus de maturation cellulaire de la protéine CFTR.
-
Classe 3 : mutations perturbant la régulation du canal chlorure.
-
Classe 4 : mutations altérant la conduction du canal chlorure.
-
Classe 5 : mutations altérant la stabilité de l'ARNm CFTR.
-
Classe 6 : mutations altérant la stabilité de la protéine mature.
La mutation la plus fréquente est la mutation « Delta F508 » (ΔF508) qui consiste en une délétion de trois nucléotides au niveau du dixième exon du gène, aboutissant à l'élimination d'un acide aminé, la phénylamine, en position 508.
Cette mutation est retrouvée dans près de deux tiers des cas avec des variations importantes selon les populations étudiées.
Seules quatre autres mutations, hors ΔF508, représentent plus de 1 % des cas, il s'agit de G542X, G551D, N1303K et W1282X1.
Toutes les autres mutations sont rares, voire exceptionnelles, uniquement décelées au sein d'une seule famille.
Les mutations les plus fréquentes dans les populations caucasiennes sont :
|
Nom de la mutation |
Fréquence |
Nom de la mutation |
Fréquence | |
|---|---|---|---|---|
|
ΔF508 |
66,0 % |
R553X |
0,7 % |
|
|
G542X |
2,4 % |
621+1G |
0,7 % |
|
|
G551D |
1,6 % |
1717-1G |
0,6 % |
|
|
N1303K |
1,3 % |
R117H |
0,3 % |
|
|
W1282X |
1,2 % |
R1162X |
0,33 % |
Il existe un certain rapport entre la mutation (le génotype) et les manifestations cliniques de la maladie (le phénotype).
La très grande variabilité des manifestations et de la gravité de cette maladie est en rapport avec les multiples mutations du gène CFTR.
La protéine CFTR
La protéine CFTR (cystic fibrosis transmembrane conductance regulator), codée par le gène CFTR, est une protéine transmembranaire de 1 480 acides aminés et dont le poids moléculaire est de 168 kD.
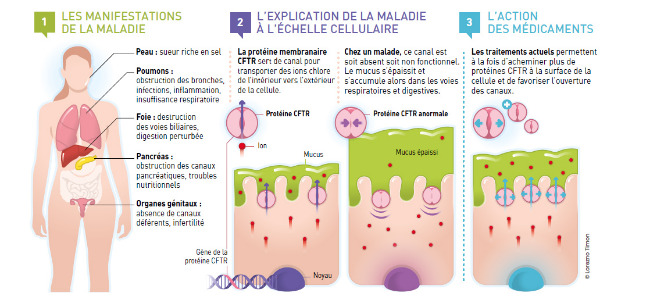
La mutation du gène de la mucoviscidose entraîne un défaut dans la synthèse de la protéine CFTR entrant dans la formation d'un canal ionique sélectif aux anions et principalement les chlorures. Outre sa fonction canal chlore, la protéine CFTR régule également d'autres canaux ce qui fait de la protéine CFTR une protéine multifonctionnelle.
SYMPTOMES et CONSEQUENCES
Le défaut de synthèse de la protéine entraîne un déficit en chlore extracellulaire, en thiocyanate et donc un défaut d'hydratation du mucus, une hyperviscosité des sécrétions épithéliales liée à une réabsorption exagérée de l'eau secondaire à la rétention cellulaire de l'ion chlore et au non passage des composés nécessaires à la défense immunitaire du poumon
L'insuffisance de fonctionnement des glandes exocrines se remarque surtout au niveau du poumon, du pancréas et du foie. Les atteintes digestives sont les plus précoces et les premières historiquement décrites.
Au niveau du pancréas
Les enzymes du pancréas, comme la lipase et la trypsine, qui servent à la digestion sont mal excrétées dans la lumière intestinale parce que les canaux pancréatiques se bouchent du fait d'une sécrétion trop épaisse et d'une fibrose importante. Les enzymes agressent alors directement le tissu pancréatique, contribuant à la fibrose. Des kystes peuvent éventuellement se former. Lorsque les fonctions pancréatiques sont significativement altérées, on parle d'insuffisance pancréatique.
Le pancréas est atteint dans sa fonction exocrine et endocrine de deux manières :
-
atteinte de la fonction exocrine : la réduction ou l'absence d'arrivée d'enzymes de digestion dans la lumière intestinale conduit à un syndrome de malabsorption pouvant avoir un retentissement sur le développement staturopondéral et pubertaire des enfants.
-
atteinte de la fonction endocrine : la destruction des cellules bêta des îlots de Langerhans conduit à un défaut de sécrétion d' insuline et un diabète insulinoprive, distinct du diabète de type 1 auto-immun.
Au niveau hépatobiliaire
La viscosité de la bile est augmentée par les dysfonctionnements de la protéine CFTR des cellules épithéliales biliaires. Les canaux qui transportent la bile vont se boucher à cause de ce liquide trop épais. La répétition de ces obstructions conduit à des phénomènes de cirrhose localisée et d'hépatomégalie.
L'hypertension dans la veine porte, les hémorragies digestives et l'insuffisance hépatocellulaire peuvent conduire à la greffe hépatique.
Au niveau du poumon
Le liquide de surface tapissant l'arbre bronchique se compose d'eau et de mucus. Les canaux CFTR servent à la sécrétion active de chlore vers ce liquide, ils interagissent avec les canaux d'absorption du sodium pour limiter leur travail. Ce mouvement de chlore entraîne un mouvement de sodium et d'eau. Cette sécrétion d'eau permet d'hydrater le liquide de surface bronchique et de lui maintenir des propriétés rhéologiques adéquates pour une clairance mucociliaire efficace. La clairance mucociliaire est le débit de liquide de surface transporté par les cellules ciliées de l'arbre bronchique. L'évacuation de ce liquide permet l'élimination des poussières et des agents infectieux vers le système digestif.
L'altération fonctionnelle des canaux CFTR entraîne une déshydratation du liquide de surface bronchique. Les modifications des propriétés du mucus, et notamment l'augmentation de sa viscosité rendant plus difficile son évacuation par les cils, conduit à une obstruction chronique des bronches ainsi qu'à la non évacuation des poussières et bactéries. De plus les propriétés antibactériennes du mucus sont diminuées. Tous ces éléments favorisent l'apparition d'une infection précoce devenant rapidement chronique associée à une réaction inflammatoire marquée.
Au fil du temps, différentes bactéries colonisent les voies respiratoires d'un même malade.
Initialement ce sont des bactéries banales telles que Haemophilus influenzae et Staphylococcus aureus qui colonisent et infectent les poumons, puis survient la colonisation à Pseudomonas aeruginosa, qui constitue un tournant dans l’évolution de la maladie respiratoire.
L'utilisation répétée d'antibiotiques mais aussi d'autres facteurs encore indéterminés et liés à la maladie elle-même, favorisent la colonisation du tractus bronchique par Pseudomonas aeruginosa généralement résistante aux antibiotiques d'utilisation courante. Cette bactérie est exceptionnelle chez les personnes indemnes de mucoviscidose.
À terme, l'inflammation et l'infection chronique entretiennent un cercle vicieux et entraînent une dégradation pulmonaire par des lésions du tissu pulmonaire conduisant à un tableau de bronchopneumopathie chronique obstructive.
La forme clinique la plus fréquente associe troubles respiratoires (bronchite chronique), troubles digestifs (stéatorrhée, constipation) et troubles de la croissance staturopondérale.
Les manifestations pulmonaires restent la cause majeure de la morbidité et de la mortalité.
L'histoire naturelle de la maladie pulmonaire est une dégradation progressive de l'état respiratoire, évoluant par poussées, débutant par des symptômes tels qu'une toux et des crachats et aboutissant à une insuffisance respiratoire grave.
Les premières manifestations respiratoires sont une toux chronique ou récurrente — initialement sèche — parfois paroxystique et pouvant entraîner des vomissements, puis grasse et éventuellement purulente, des crachats et des difficultés expiratoires qui surviennent lorsque la fonction respiratoire se détériore.
Des épisodes de bronchites chroniques surviennent chez l'enfant et entraînent progressivement des lésions de l'arbre bronchique.
Une fois l'atteinte respiratoire bien installée, les malades peuvent présenter des douleurs thoraciques, des pneumopathies récurrentes, des formes atypiques d'asthme et un wheezing récurrent.
Des épisodes de pneumothorax ou d'hémoptysies peuvent engager le pronostic vital.
Les lésions des poumons s'aggravent progressivement aboutissant à une insuffisance respiratoire majeure.
Manifestations digestives
Manifestations intestinales
Les manifestations surviennent dès la période néonatale sous forme d'une occlusion intestinale aiguë qui survient chez 10 à 20 % des enfants porteurs de la maladie et peut être diagnostiqué lors de l'échographie morphologique. Cette occlusion intestinale aiguë néonatale basse qui se traduit chez le nouveau né par l'absence d'émission de méconium associé à des vomissements et à des ballonnements. Le lavement permet l'évacuation d'un méconium obstructif visqueux
Le reflux gastro-oesophagien est fréquent chez le nourrisson, parfois majoré par la toux chronique.
Le prolapsus rectal est également plus fréquent chez les nourrissons mucoviscidosiques et justifie la réalisation d'un test de la sueur.
Manifestations hépato biliaires
Éléments anatomiques de l'arbre biliaire
L'atteinte hépato-biliaire de la mucoviscidose est fréquente mais n'évolue vers la cirrhose biliaire que dans 10 à 15 % des cas.
Elle se traduit par une élévation de la viscosité de la bile puis par une obstruction à l'évacuation de cette dernière dans les canaux hépatiques vers la vésicule biliaire.
La conséquence est l'apparition d'une coloration jaune de la peau et des muqueuses qui est le reflet de l'élévation de la bilirubine non conjuguée dans le sang.
Manifestations pancréatiques
L'insuffisance pancréatique exocrine est présente chez la grande majorité des patients, dans environ 90 % des cas.
Elle est responsable d'un défaut d'absorption des graisses (acides gras essentiels), des protéines et des vitamines liposolubles (A, D, E, K et parfois hydrosoluble comme B12).
Parmi les autres manifestations de lamalabsorption, on retrouve une insuffisance de croissance, un retard pubertaire, une anémie, des troubles carentiels (en vitamines, oligo-éléments et minéraux).
On peut voir parfois l'apparition de pancréatite aiguë ou chronique, plus fréquemment chez les patients avec une fonction pancréatique conservée.
Manifestations génitales
Les manifestations génitales de la mucoviscidose posent un problème de plus en plus important avec l'amélioration de la survie et le désir d'enfant des patients.
Chez la femme on retrouve une hypofertilité due à des modifications de la glaire cervicale, néanmoins les grossesses de femmes mucoviscidosiques ne sont plus rares.
Chez l' homme on retrouve une stérilité par obstruction atrésie bilatérale des canaux déférents. Cette anomalie entraîne une azoospermie ou oligospermie sévère (moins de 5 millions de spermatozoïdes par ml). La composition du sperme est aussi modifiée :
|
Normal |
Mucoviscidose | |
|---|---|---|
|
pH |
>8 |
<7 |
|
Acide citrique |
400-1 500 /100 ml |
> 2 000 /100 ml |
|
Phosphatase acide |
140-290 |
760-1 140 |
|
Fructose |
250-720 /100 ml |
30–80 /100 ml |
Autres manifestations
Les affections rhinosinusiennes sont fréquentes. La qualité du mucus dans les sinus est la même que celle des bronches. Une inflammation et une infection chronique entraîne une sinusite chronique et secondairement une polypose nasale.
Les désordres hydroélectrolytiques avec déshydratation par perte de sel peuvent apparaitre lors d'efforts ou de forte chaleur.
Une Cardiomyopathie probablement d'origine carentielle peut être décrite ainsi qu'une hypertension artérielle pulmonaire consécutive à un cœur pulmonaire chronique.


{kind=link}